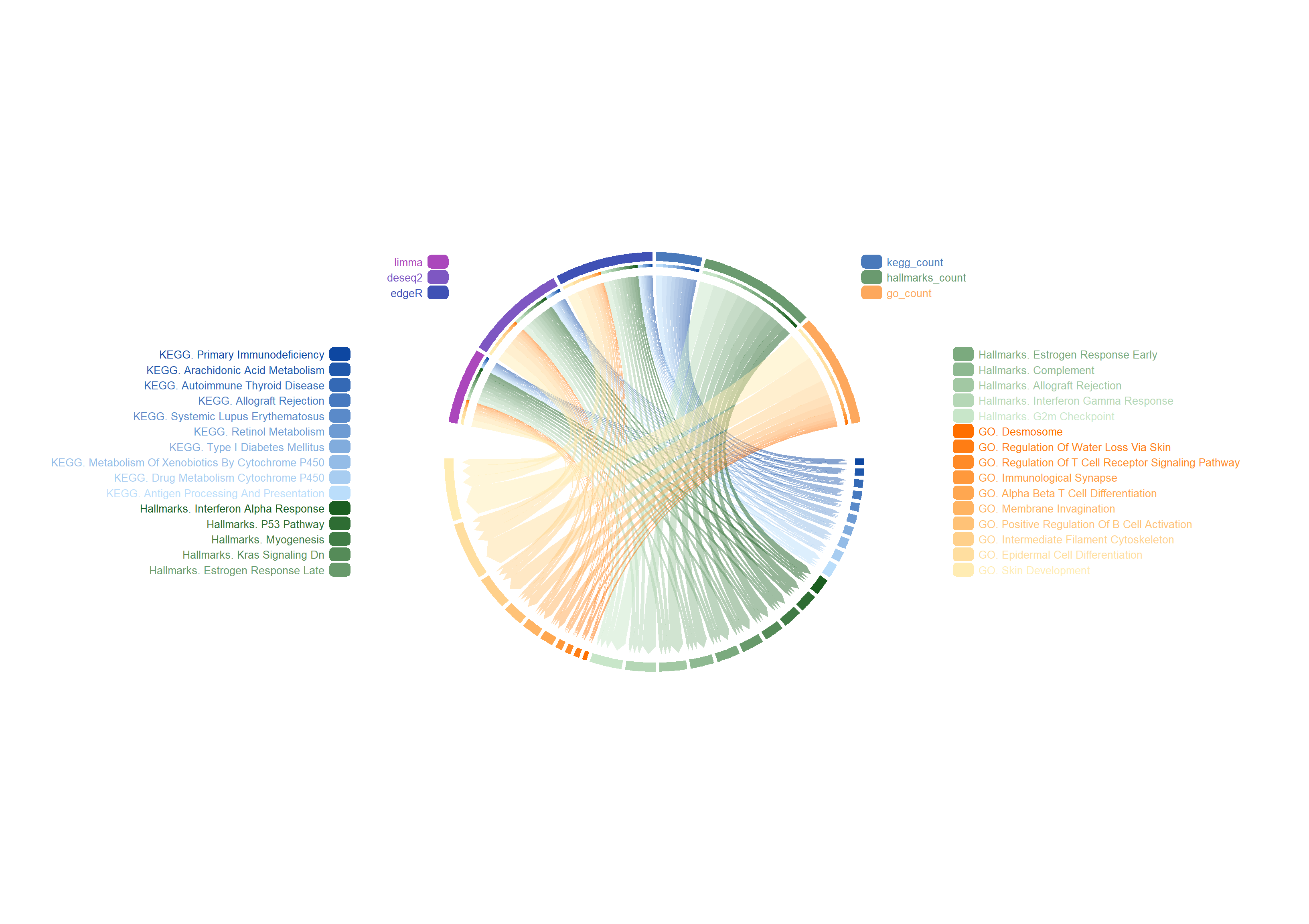
Integrated visualization of different samples, methods, and pathways: Includes specific naming annotations, sorting by the number of enriched genes, and unique arrangement without grouping.
result <-adjust_export_pathway(fgseaRes =deseq2_hallmarks_fgseaRes, nTop =10)deseq2_hallmarks_UpDown_topPathways <- result$combinedPathwaysdeseq2_hallmarks_fgseaRes <- result$fgseaRes
result <-adjust_export_pathway(fgseaRes =deseq2_go_fgseaRes, nTop =100)deseq2_go_UpDown_topPathways <- result$combinedPathwaysdeseq2_go_fgseaRes <- result$fgseaRes
result <-adjust_export_pathway(fgseaRes =deseq2_kegg_fgseaRes, nTop =10)deseq2_kegg_UpDown_topPathways <- result$combinedPathwaysdeseq2_kegg_fgseaRes <- result$fgseaRes
14.4.2 edgeR
result <-adjust_export_pathway(fgseaRes =edgeR_hallmarks_fgseaRes, nTop =10)edgeR_hallmarks_UpDown_topPathways <- result$combinedPathwaysedgeR_hallmarks_fgseaRes <- result$fgseaRes
result <-adjust_export_pathway(fgseaRes =edgeR_go_fgseaRes, nTop =100)edgeR_go_UpDown_topPathways <- result$combinedPathwaysedgeR_go_fgseaRes <- result$fgseaRes
result <-adjust_export_pathway(fgseaRes =edgeR_kegg_fgseaRes, nTop =10)edgeR_kegg_UpDown_topPathways <- result$combinedPathwaysedgeR_kegg_fgseaRes <- result$fgseaRes
14.4.3 limma
result <-adjust_export_pathway(fgseaRes =limma_hallmarks_fgseaRes, nTop =10)limma_hallmarks_UpDown_topPathways <- result$combinedPathwayslimma_hallmarks_fgseaRes <- result$fgseaRes
result <-adjust_export_pathway(fgseaRes =limma_go_fgseaRes, nTop =100)limma_go_UpDown_topPathways <- result$combinedPathwayslimma_go_fgseaRes <- result$fgseaRes
result <-adjust_export_pathway(fgseaRes =limma_kegg_fgseaRes, nTop =10)limma_kegg_UpDown_topPathways <- result$combinedPathwayslimma_kegg_fgseaRes <- result$fgseaRes
14.5 common_elements
# Use Reduce and intersect to calculate the intersection of all vectorskegg_UpDown_common_elements <-Reduce(intersect, list(limma_kegg_UpDown_topPathways, edgeR_kegg_UpDown_topPathways, deseq2_kegg_UpDown_topPathways))# Print the intersection resultsprint(kegg_UpDown_common_elements)
[1] "Primary Immunodeficiency"
[2] "Allograft Rejection"
[3] "Graft Versus Host Disease"
[4] "Autoimmune Thyroid Disease"
[5] "Type I Diabetes Mellitus"
[6] "Systemic Lupus Erythematosus"
[7] "Antigen Processing And Presentation"
[8] "Arachidonic Acid Metabolism"
[9] "Metabolism Of Xenobiotics By Cytochrome P450"
[10] "Drug Metabolism Cytochrome P450"
[11] "Retinol Metabolism"
# Use Reduce and intersect to calculate the intersection of all vectorsgo_UpDown_common_elements <-Reduce(intersect, list(limma_go_UpDown_topPathways, edgeR_go_UpDown_topPathways, deseq2_go_UpDown_topPathways))# Print the intersection resultsprint(go_UpDown_common_elements)
[1] "Mhc Protein Complex"
[2] "Mhc Protein Complex Binding"
[3] "Peptide Antigen Binding"
[4] "Lumenal Side Of Endoplasmic Reticulum Membrane"
[5] "Tolerance Induction"
[6] "T Cell Receptor Complex"
[7] "Positive Regulation Of Interleukin 2 Production"
[8] "Immunoglobulin Complex Circulating"
[9] "Regulatory T Cell Differentiation"
[10] "Lumenal Side Of Membrane"
[11] "Immunoglobulin Receptor Binding"
[12] "Positive T Cell Selection"
[13] "Regulation Of Natural Killer Cell Mediated Immunity"
[14] "T Cell Mediated Cytotoxicity"
[15] "Regulation Of T Cell Receptor Signaling Pathway"
[16] "Immunological Synapse"
[17] "Cellular Defense Response"
[18] "T Cell Selection"
[19] "Phagocytosis Recognition"
[20] "Negative Regulation Of Viral Genome Replication"
[21] "Positive Regulation Of Cell Killing"
[22] "Pigment Biosynthetic Process"
[23] "Immunoglobulin Complex"
[24] "Regulation Of B Cell Proliferation"
[25] "Positive Regulation Of Alpha Beta T Cell Activation"
[26] "Regulation Of Antigen Receptor Mediated Signaling Pathway"
[27] "Regulation Of Complement Activation"
[28] "Positive Regulation Of Interferon Gamma Production"
[29] "Regulation Of Leukocyte Mediated Cytotoxicity"
[30] "Tertiary Granule Membrane"
[31] "B Cell Proliferation"
[32] "Regulation Of Cell Killing"
[33] "Specific Granule Membrane"
[34] "Humoral Immune Response Mediated By Circulating Immunoglobulin"
[35] "Positive Regulation Of T Cell Proliferation"
[36] "B Cell Receptor Signaling Pathway"
[37] "Antigen Binding"
[38] "Regulation Of Alpha Beta T Cell Activation"
[39] "Regulation Of Humoral Immune Response"
[40] "Complement Activation"
[41] "Membrane Invagination"
[42] "Positive Regulation Of B Cell Activation"
[43] "Alpha Beta T Cell Differentiation"
[44] "Blood Microparticle"
[45] "Positive Regulation Of Leukocyte Mediated Immunity"
[46] "Epithelial Cell Proliferation"
[47] "Endopeptidase Activity"
[48] "Fatty Acid Metabolic Process"
[49] "Epidermis Development"
[50] "Skin Development"
[51] "Epidermal Cell Differentiation"
[52] "Contractile Fiber"
[53] "Fat Cell Differentiation"
[54] "Keratinocyte Differentiation"
[55] "Hormone Metabolic Process"
[56] "Serine Hydrolase Activity"
[57] "Intermediate Filament Cytoskeleton"
[58] "Keratinization"
[59] "Response To Xenobiotic Stimulus"
[60] "Intermediate Filament"
[61] "Striated Muscle Cell Development"
[62] "Primary Alcohol Metabolic Process"
[63] "Cellular Component Assembly Involved In Morphogenesis"
[64] "Skin Epidermis Development"
[65] "Desmosome"
[66] "Regulation Of Water Loss Via Skin"
[67] "Aromatase Activity"
[68] "Cell Cell Junction"
[69] "Regulation Of Systemic Arterial Blood Pressure"
[70] "Epithelial Cell Development"
[71] "Oxidoreductase Activity Acting On Paired Donors With Incorporation Or Reduction Of Molecular Oxygen"
[72] "Lipid Catabolic Process"
[73] "Cellular Modified Amino Acid Metabolic Process"
[74] "Heart Morphogenesis"
[75] "Cell Cell Junction Organization"
[76] "Cardiac Muscle Tissue Development"
# Use Reduce and intersect to calculate the intersection of all vectorshallmarks_UpDown_common_elements <-Reduce(intersect, list(limma_hallmarks_UpDown_topPathways, edgeR_hallmarks_UpDown_topPathways, deseq2_hallmarks_UpDown_topPathways))# Print the intersection resultsprint(hallmarks_UpDown_common_elements)
# Randomly select 10 pathways from the provided list, ensuring that each pathway name does not exceed 10 words.hallmarks_selected_pathways <-selectPathways(hallmarks_UpDown_common_elements, max_words =10, num_select =10)print(hallmarks_selected_pathways)
colnames(limma_hallmarks_filtered_10fgseaRes) <-c("pathway", "limma")# Use full_join to merge dataframeshallmarks_combined_df <- deseq2_hallmarks_filtered_10fgseaRes %>%full_join(edgeR_hallmarks_filtered_10fgseaRes, by ="pathway") %>%full_join(limma_hallmarks_filtered_10fgseaRes, by ="pathway")# View the merged dataframeprint(hallmarks_combined_df)
# Add a prefix "hallmarks." to the pathway columnhallmarks_combined_df <- hallmarks_combined_df %>%mutate(pathway =paste("Hallmarks.", pathway))# Print the modified dataframe to view resultsprint(hallmarks_combined_df)
pathway genecount
<char> <int>
1: Epidermal Cell Differentiation 117
2: Membrane Invagination 42
3: Regulation Of Water Loss Via Skin 14
4: Skin Development 136
5: Alpha Beta T Cell Differentiation 36
6: Regulation Of T Cell Receptor Signaling Pathway 17
7: Positive Regulation Of B Cell Activation 46
8: Immunological Synapse 21
9: Desmosome 12
10: Intermediate Filament Cytoskeleton 48
colnames(limma_go_filtered_10fgseaRes) <-c("pathway", "limma")library(dplyr)# Use full_join to merge dataframesgo_combined_df <- deseq2_go_filtered_10fgseaRes %>%full_join(edgeR_go_filtered_10fgseaRes, by ="pathway") %>%full_join(limma_go_filtered_10fgseaRes, by ="pathway")# View the merged dataframeprint(go_combined_df)
pathway deseq2 edgeR limma
<char> <int> <int> <int>
1: Epidermal Cell Differentiation 209 204 117
2: Membrane Invagination 60 58 42
3: Regulation Of Water Loss Via Skin 18 18 14
4: Skin Development 224 216 136
5: Alpha Beta T Cell Differentiation 43 44 36
6: Regulation Of T Cell Receptor Signaling Pathway 19 19 17
7: Positive Regulation Of B Cell Activation 76 68 46
8: Immunological Synapse 21 21 21
9: Desmosome 14 14 12
10: Intermediate Filament Cytoskeleton 128 131 48
# Add a prefix "GO." to the pathway columngo_combined_df <- go_combined_df %>%mutate(pathway =paste("GO.", pathway))# Print the modified dataframe to view resultsprint(go_combined_df)
pathway deseq2 edgeR limma
<char> <int> <int> <int>
1: GO. Epidermal Cell Differentiation 209 204 117
2: GO. Membrane Invagination 60 58 42
3: GO. Regulation Of Water Loss Via Skin 18 18 14
4: GO. Skin Development 224 216 136
5: GO. Alpha Beta T Cell Differentiation 43 44 36
6: GO. Regulation Of T Cell Receptor Signaling Pathway 19 19 17
7: GO. Positive Regulation Of B Cell Activation 76 68 46
8: GO. Immunological Synapse 21 21 21
9: GO. Desmosome 14 14 12
10: GO. Intermediate Filament Cytoskeleton 128 131 48
pathway genecount
<char> <int>
1: Arachidonic Acid Metabolism 19
2: Systemic Lupus Erythematosus 26
3: Primary Immunodeficiency 16
4: Retinol Metabolism 19
5: Metabolism Of Xenobiotics By Cytochrome P450 22
6: Drug Metabolism Cytochrome P450 24
7: Antigen Processing And Presentation 35
8: Type I Diabetes Mellitus 26
9: Autoimmune Thyroid Disease 25
10: Allograft Rejection 24
colnames(limma_kegg_filtered_10fgseaRes) <-c("pathway", "limma")library(dplyr)# Use full_join to merge dataframeskegg_combined_df <- deseq2_kegg_filtered_10fgseaRes %>%full_join(edgeR_kegg_filtered_10fgseaRes, by ="pathway") %>%full_join(limma_kegg_filtered_10fgseaRes, by ="pathway")# View the merged dataframeprint(kegg_combined_df)
# Add a prefix "KEGG." to the pathway columnkegg_combined_df <- kegg_combined_df %>%mutate(pathway =paste("KEGG.", pathway))# Print the modified dataframe to view resultsprint(kegg_combined_df)
# Calculate the total number of genes across different samples/methods for the same pathway# Add 'hallmarks_count' column to the dataframehallmarks_combined_count_df <- hallmarks_combined_df %>%mutate(hallmarks_count =rowSums(select(., deseq2, edgeR, limma), na.rm =TRUE))hallmarks_combined_count_df <- hallmarks_combined_count_df %>%arrange(hallmarks_count) # Sort in ascending order by defaultprint(hallmarks_combined_count_df)
# Calculate the total number of genes across different samples/methods for the same pathway# Add 'go_count' column to the dataframego_combined_count_df <- go_combined_df %>%mutate(go_count =rowSums(select(., deseq2, edgeR, limma), na.rm =TRUE))go_combined_count_df <- go_combined_count_df %>%arrange(go_count) # Sort in ascending order by defaultprint(go_combined_count_df)
# Calculate the total number of genes across different samples/methods for the same pathway# Add 'kegg_count' column to the dataframekegg_combined_count_df <- kegg_combined_df %>%mutate(kegg_count =rowSums(select(., deseq2, edgeR, limma), na.rm =TRUE))kegg_combined_count_df <- kegg_combined_count_df %>%arrange(kegg_count) # Sort in ascending order by defaultprint(kegg_combined_count_df)
# Use pivot_longer to convert from wide to long formatkegg_long_df <- kegg_combined_count_df %>%pivot_longer(cols =c(limma, deseq2, edgeR, kegg_count),names_to ="method/group",values_to ="genecount" )# Print the transformed long format dataframeprint(kegg_long_df)
# Use pivot_longer to convert from wide to long formatgo_long_df <- go_combined_count_df %>%pivot_longer(cols =c(limma, deseq2, edgeR, go_count),names_to ="method/group",values_to ="genecount" )# Print the transformed long format dataframeprint(go_long_df)
# A tibble: 40 × 3
pathway `method/group` genecount
<chr> <chr> <dbl>
1 GO. Desmosome limma 12
2 GO. Desmosome deseq2 14
3 GO. Desmosome edgeR 14
4 GO. Desmosome go_count 40
5 GO. Regulation Of Water Loss Via Skin limma 14
6 GO. Regulation Of Water Loss Via Skin deseq2 18
7 GO. Regulation Of Water Loss Via Skin edgeR 18
8 GO. Regulation Of Water Loss Via Skin go_count 50
9 GO. Regulation Of T Cell Receptor Signaling Pathway limma 17
10 GO. Regulation Of T Cell Receptor Signaling Pathway deseq2 19
# ℹ 30 more rows
# Use pivot_longer to convert from wide to long formathallmarks_long_df <- hallmarks_combined_count_df %>%pivot_longer(cols =c(limma, deseq2, edgeR, hallmarks_count),names_to ="method/group",values_to ="genecount" )# Print the transformed long format dataframeprint(hallmarks_long_df)
# Use rbind to merge these three dataframesall_combined_df <-rbind(kegg_long_df, hallmarks_long_df, go_long_df)# Print the merged dataframe to check the resultsprint(all_combined_df)